April 9, 2012 — Thoratec and the U.S. Food and Drug Adminstration (FDA) announced a recent Class 1 recall of the HeartMate II left ventricular assist device (LVAD) because detachment of the bend relief from its intended position around the proximal outflow graft may allow the graft to kink or deform, resulting in reduction of blood flow. The kink also may cause pump/graft thrombosis or perforation of the outflow graft. Additionally, the metal end of the bend relief may be sharp and cause erosion and cutting of the outflow graft.
Model numbers affected are: 103393, 103695, 104692, 104911, 104912.
The HeartMate II LVAS is indicated for use as a bridge to transplantation in heart transplant candidates at risk of imminent death from non-reversible left ventricular failure. It is also indicated for use in patients with New York Heart Association (NYHA) Class IIB of IV end-stage left-ventricular failure who have received optimal medical therapy for at least 45 of the last 60 days and who are not candidates for heart transplantation.
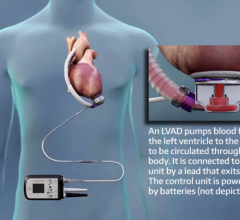
The HeartMate II blood pump delivers blood from the dysfunctional left ventricle of the heart to the rest of the body. It is an axial flow rotary ventricular assist system and can generate blood flows up to 10 liters per minute (one beat per minute). The device is intended to be used both inside and outside the hospital (such as at home), or during transportation of ventricular assist device patients by ground ambulance, airplane or helicopter.
The device labeling has been revised to provide instructions on how to verify the bend relief is fully engaged with the sealed outflow graft at the time of implant, and new caution statements regarding the bend relief connection are included. Clinicians have been instructed to follow the revised instructions that clarify the recommended procedure for securing the bend relief to the outflow graft.
The company initiated the voluntary notification to clinicians Feb. 23, 2012. Thoratec contacted all hospitals to whom HeartMate II sealed outflow grafts had been distributed. The company's voluntary notification was intended to provide information to clinicians, including a clarification of the instructions for use, and did not involve the return of any product. Subsequently, the company received acknowledgment forms from surgeons at all such U.S. hospitals, verifying that clinicians reviewed the notification and understand the information provided.
Thoratec anticipates no material financial impact from this action.
For more information: www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm298710.htm

 June 19, 2024
June 19, 2024