January 11, 2018 — The U.S. Food and Drug Administration (FDA) announced that Edwards LifeSciences is recalling its Certitude Delivery System due to a molding overflow defect in the button valve within the loader. The overflow material could detach during placement of the delivery system and potentially embolize into the patient, according to the FDA. Such an embolism could obstruct blood flow to critical organs, leading to serious injury and/or a need to surgically extract the overflow material from the patient. In dire situations, severe neurologic, cardiac, limb, renal or gastrointestinal injury may result.
The recall encompasses select lot numbers between 60677270 and 60990824, with manufacturing dates between Nov. 22, 2016 to July 10, 2017, and distribution dates between Jan. 9, 2017 to July 17, 2017.
On July 21, 2017, Edwards LifeSciences sent affected customers a "Recall Notification Letter" informing them of the device's risks. In the letter, Edwards LifeSciences directed customers to:
- Complete the "Acknowledgement Form" that accompanied the Recall Notification Letter.
- Check all inventory for affected models of the Certitude Delivery System.
- Return the "Acknowledgement Form" and all affected models of the Certitude Delivery System to Edwards LifeSciences as indicated in the Recall Notification Letter.
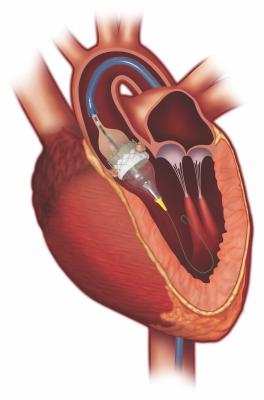
The Edwards LifeSciences Certitude Delivery System is used for delivery of the Edwards Sapien 3 transcatheter heart valve (THV), typically used during a transcatheter aortic valve replacement (TAVR).
The system includes a balloon catheter that expands a compressed (crimped) THV, a loader that delivers the THV through the guiding tube and extension tubing. During a procedure, the physician will first crimp the THV onto the balloon of the Certitude Delivery System. The system is then inserted into the body, usually during a transapical (inserted through small incision under left breast) or transaortic (inserted through small incision in the top right side of the chest) approach. The THV is then deployed through the guiding tube to the site of the native stenotic aortic valve, where it is expanded and fixed in place.
Customers with questions regarding this recall may contact Edwards Customer Service at 1-800-424-3278, from 6:00 AM to 4:30 PM (Pacific Time).
Healthcare professionals and patients are encouraged to report adverse events or side effects related to the use of these products to the FDA's MedWatch Safety Information and Adverse Event Reporting Program:
- Complete and submit the report Online: www.fda.gov/MedWatch/report
- Download form or call 1-800-332-1088 to request a reporting form, then complete and return to the address on the pre-addressed form, or submit by fax to 1-800-FDA-0178
For more information: www.fda.gov

 November 14, 2025
November 14, 2025 







